
随着计算化学和人工智能技术的快速发展,量子化学计算(Quantum Chemistry Computation)正逐步成为创新药物研发的重要工具。基于密度泛函理论(DFT)和从头算(ab initio)方法的分子电子结构计算,能够精准解析小分子与靶蛋白的相互作用,为优化先导化合物提供高分辨率的结构和能量信息。近年来,量子化学计算在以下几个方面展现出强大的应用潜力:
1) 结合自由能计算与能量分解
相比传统的分子对接和分子动力学模拟,量子化学计算可以通过SAPT(对称适应微扰理论)等方法更精确地解析氢键、π-π相互作用、范德华力等非共价相互作用,为药物优化提供定量的能量分解数据。
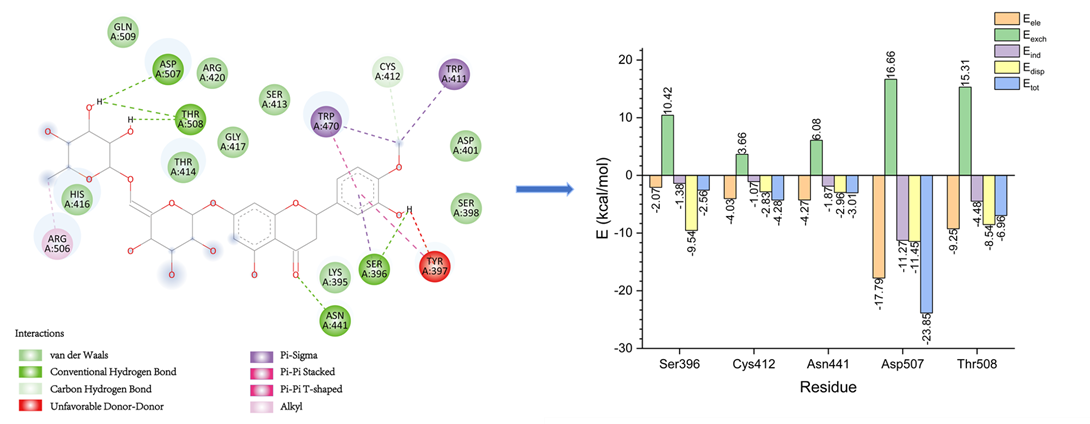
小分子和蛋白相互作用SAPT能量分解
2) 共价抑制剂设计
随着共价药物的崛起,量子化学计算在反应路径、活性位点的电荷分布及反应势能面(PES)预测等方面发挥关键作用。例如,通过DFT计算可以评估亲电试剂与靶蛋白关键残基(如Cys、Ser)的成键活性,优化共价抑制剂的结构。
3)量子机器学习(Quantum Machine Learning, QML)
结合量子化学计算和机器学习,研究者可以建立高精度的能量预测模型,大幅提升虚拟筛选的准确性和计算效率,尤其适用于大规模化合物库的筛选和活性预测。
4) 溶剂化效应与质子转移研究
许多药物分子在生理环境中的活性受pKa和溶剂效应影响,量子化学计算能够通过连续介质模型(PCM)或分子簇模型模拟溶剂效应,辅助优化化合物的理化性质,提高成药性。
5) 金属离子依赖靶点的药物设计
对于涉及金属离子(如Zn²⁺、Mg²⁺、Fe³⁺)的蛋白靶点,量子化学计算可以模拟金属配位环境,预测抑制剂的结合模式,为金属酶抑制剂和螯合剂的设计提供理论依据。
经典案例分享:QM计算揭示降血糖药物共价结合靶点机制
传统药物通过与靶标之间的非键相互作用实现结合,这些作用主要包括静电相互作用、氢键、范德华力以及疏水效应等;而共价药物在此基础上会进一步与靶标形成强化学键(如共价键),从而显著增强结合紧密性。然而,共价结合过程往往涉及电子转移和化学键的断裂/重组,这超出了传统分子力学方法的描述能力。本研究以DPP-4抑制剂Vildagliptin(VIL)和Saxagliptin(SAX)为研究对象,这两种药物虽结构相似,但其与靶点的结合行为却截然不同:实验观察到VIL表现出伪不可逆抑制特性,而SAX则通过可逆共价修饰实现更高活性。传统理论模型难以解释这种差异,而基于量子力学/分子力学(QM/MM)的分子动力学模拟则揭示了其本质原因:QM方法通过高精度电子结构计算,发现VIL的水解途径主导其解离动力学,而SAX的吡咯烷环4,5-亚甲基取代基通过稳定过渡态使其共价键更易逆转。这一案例不仅验证了QM/MM在解析共价药物复杂机制中的独特优势,更展现了其在揭示电子转移、键合细节等微观过程方面的不可替代性。
运用量子力学-分子力学组合(QM/MM)的多尺度模拟方法,详尽地阐明了治疗糖尿病的共价药物维格列汀(VIL)和沙格列汀(SAX)与靶标DPP-4作用的生物化学机制,并从酶催化的角度揭示了导致两种药物活性差异的原因(图1)。
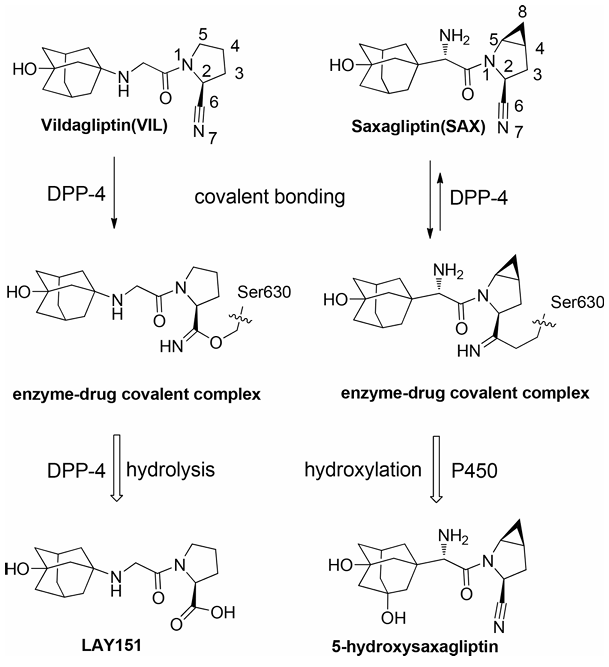
图1. 两种共价药物在靶标DPP-4活性口袋中可能经历的生物化学反应过程
传统非共价药物的活性主要由动力学常数k1和k-1决定,研究中通常用IC50值(达到50%抑制效果时抑制剂的浓度)来表征;而共价药物的活性通常不能用IC50值来衡量,其活性主要由k2、k-2和k3决定。当共价药物不能被靶标自身代谢时,也即k3不存在或很小时:如果k2远远大于k-2,药物与靶标结合是不可逆的,药物-靶标停留时间最长,同时也可能带来大的毒性;如果k-2与k2接近,药物与靶标结合是可逆的,其药物-靶标停留时间主要由k-2决定。当共价药物能被靶标自身代谢时,药物与靶标结合是伪不可逆的,其药物-靶标停留时间主要由k3决定。(如图2所示)
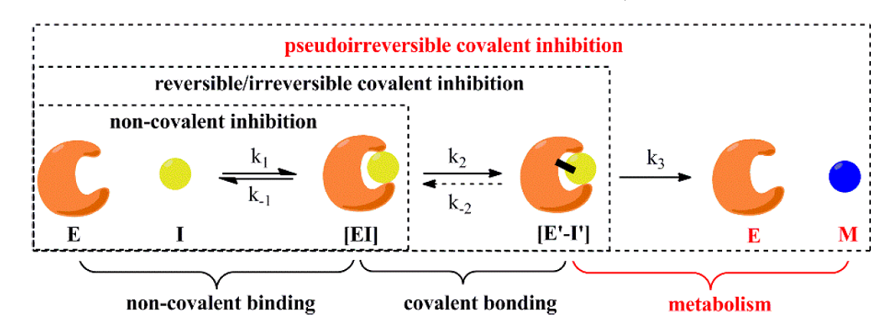
图2. 共价药物与靶标可能的作用模式
本文主要通过QM/MM MD伞形采样获得了这两种药物(VIL和SAX)与靶标DPP-4共价结合及代谢过程的自由能曲线(如图3所示)。模拟结果预示了VIL为伪不可逆共价抑制剂,而SAX为可逆共价抑制剂,与药物代谢实验相吻合,很好地解释了它们的药效差异。更重要的是,作者通过动态结构分析找出了导致这两种共价药物性质差异的潜在原因。在水解(代谢)反应中,反应中心由平图结构变成四面体结构,这将导致-CNH基团顺时针转动。如图4所示,在VIL中-CNH基团的转动是允许的;而在SAX中,三元环的空间立体排斥阻止了-CNH基团的旋转,从而导致水解反应不能发生,也即SAX不能被DPP-4自身代谢。因此,文章从酶反应动力学层面揭示了药物作用机理及活性差异的来源,对共价药物设计具有启发意义。
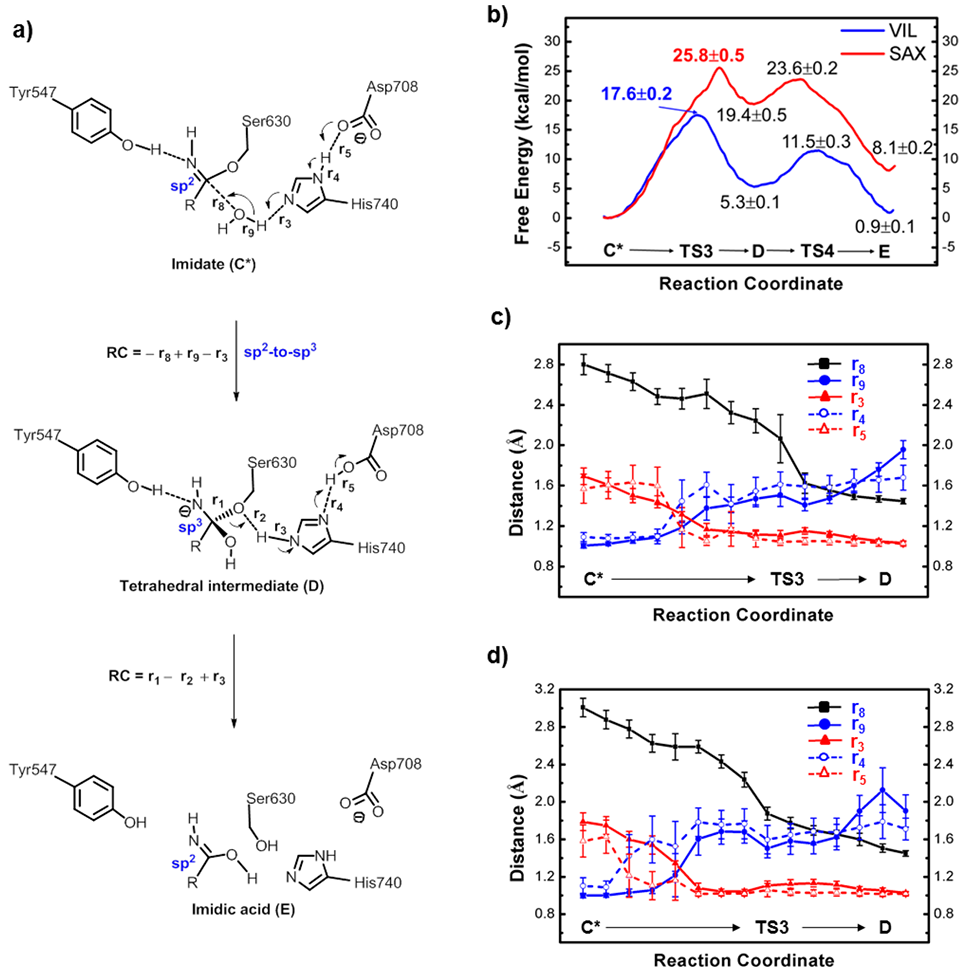
图3. 共价结合及代谢过程的自由能曲线
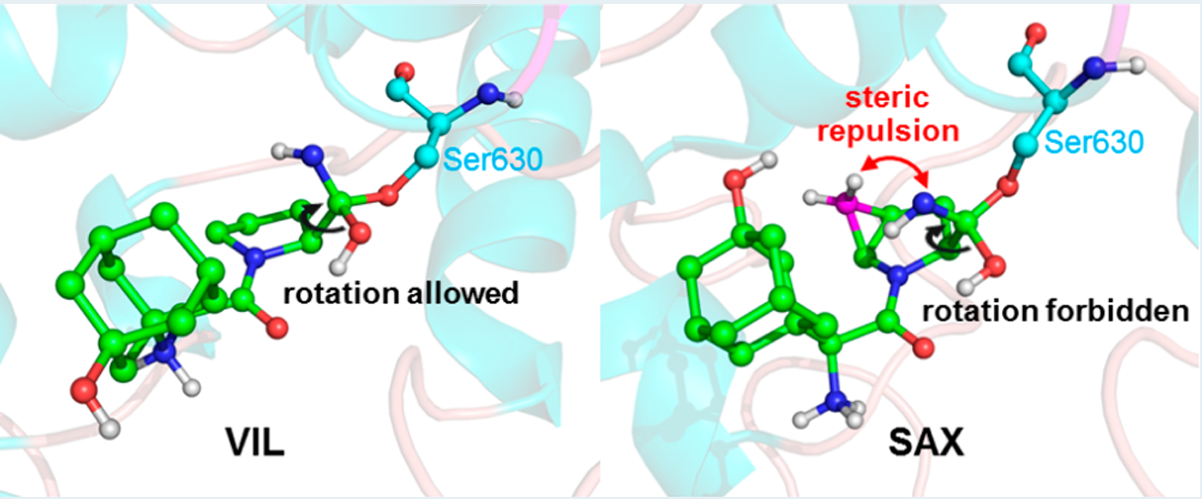
图4. 两种共价药物水解过程的代表结构
量子化学计算通过高精度电子结构分析,为药物设计提供了从分子尺度到电子尺度的深层洞见。在传统分子力学难以覆盖的共价药物领域,其优势尤为显著:一方面,基于QM/MM的模拟方法能够解析共价键形成/断裂的动态过程(如DPP-4抑制剂Vildagliptin和Saxagliptin的催化路径差异),揭示电子转移、过渡态稳定性和溶剂化效应等关键机制;另一方面,通过自由能计算与构象分析,可精准预测药物-靶标结合的持久性、代谢稳定性及脱靶风险,指导共价修饰位点优化与先导化合物迭代。此外,量子化学与机器学习、化学生物学实验的交叉融合,正推动药物设计从“经验驱动”向“计算指导实验”的范式转变。未来,随着计算效率的提升与多尺度建模技术的发展,量子化学有望在靶向不可成药蛋白、精准调控动态蛋白互作等前沿领域发挥更大作用,为下一代药物研发注入核心驱动力。
参考文献
Wang, Y.,et al. Covalent Inhibition Mechanism of Antidiabetic Drugs-Vildagliptin vs Saxagliptin. ACS Catal. 2019, 9, 2292-2302.


 沪公网安备 31011002003500
沪公网安备 31011002003500